qpc常见问题(qpc失败的原因分析)
导语:qPCR 解决方案
1. qPCR原理
利用实时荧光信号的变化监测整个PCR进程,对未知模板进行定量分析的方法,主要分为染料法荧光定量检测和探针法荧光定量检测。其特点有操作简单、快速方便、灵敏度高、重复性好、污染率低等,不仅应用于基因表达解析,还被广泛应用于SNP分型解析、病毒和病原菌的检测、物种鉴定、转基因食品的定量分析等诸多领域。
基线期:PCR反应早期,循环数较少,产物累积较少,产生的荧光的水平不能与荧光本底背景明显区分。
平台期:PCR反应过程中产生的DNA拷贝数是以指数方式增加的,随着反应循环数的增加,最终PCR反应不再以指数方式生成模板,从而进入平台期。
Ct值(Cycle threshold):表示每个PCR反应管内荧光信号到达设定的域值时所经历的循环数。Ct值与起始模板数的对数值之间存在线性关系,因此,通过已知浓度样品的Ct值和未知浓度样品的Ct值,可以计算求出未知样品的起始模板量。
熔解曲线(Melt curve):检测是荧光强度与温度的变化,可以用来确定不同的反应产物,包括非特异性产物。
决定系数(r Squared):反映标准曲线的直线性,越接近于1说明直线性越好,定量越准确。
斜率(Slope):反映PCR扩增效率,-0.25~-0.33(根据不同装置数值不同)。扩增效率(Efficiency):理想情况下扩增效率应在0.8﹤E﹤1.2。
2. 染料法荧光定量和探针法荧光定量的比较

3. 绝对定量与相对定量
绝对定量:使用已知拷贝数的绝对标准品,制作标准曲线,对未知样品的绝对量(拷贝数)进行测定的方法;通常应用于在病毒、细菌、衣原体、支原体及转基因食品的检测中应用广泛。
作为标准品应尽量选择与实际检测样品结构近似的样品,以基因组DNA为起始材料时需选择基因组DNA作为标准品;进行mRNA表达解析时需选择表达目的基因的总RNA或逆转获得的cDNA作为标准品,但在绝对定量推荐使用RNA梯度稀释后进行逆转录和qPCR的标准曲线制作。
相对定量:是分别测定目的基因和参比基因(内参基因)的量,再求出对于参比基因的目的基因的相对量,进行样品间相对量的比较。主要用于检测细胞mRNA表达量的变化;比较不同组织的mRNA表达差异;验证基因芯片、基因过表达或siRNA干扰的实验结果。
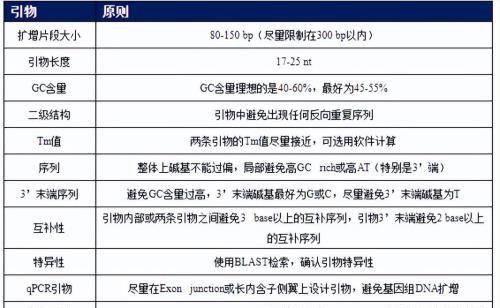
4. 荧光定量引物设计原则

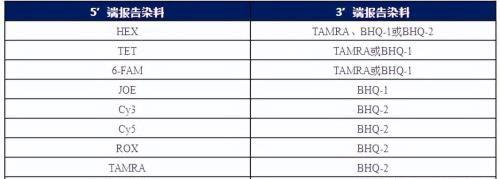
5. TaqMan探针常用荧光-淬灭对
探针法通常是使用5’端带有荧光物质(如:FAM等), 3’端带有淬灭物质(如:TAMRA、BHQ等)的探针进行荧光检测的方法。
6. 如何选择Two Step RT-PCR和One Step RT-PCR?
基因表达解析等绝大部分RT-PCR,建议选择Two Step RT-PCR。可选择随机引物、OligodT或基因特异性引物进行逆转录反应,然后再将反转录反应液(cDNA)或稀释后作为模板进行RT-qPCR。
RNA病毒检测等RT-PCR,建议选择One Step RT-PCR,反转录反应引物只能使用基因的特异性PCR下游引物。其逆转录反应和PCR反应在同一反应体系内进行,操作简便,降低污染。但这一反应体系并非逆转录反应和PCR反应的最佳反应条件,因此,One Step RT-PCR要比Two Step RT-PCR反应效率低。
7. 总RNA中混有基因组DNA时处理方法
Ø 引物设计在Exon junction或长内含子侧翼上,避免基因组DNA扩增。引物选择跨越较长的内含子,在这个内含子两侧的外显子上分别设计上、下游引物。
Ø 使用DNase I处理去除基因组DNA污染:使用常规方法提取Total RNA后,再使用DNase I处理。
Ø 可选用使用ExonScript RT SuperMix with dsDNase 逆转录试剂盒,可以高效去除残留的基因组DNA。去除基因组DNA和逆转录反应在同一管内完成。
8. 荧光定量实验成功的关键要素
Ø 总RNA需通过电泳和OD分析确认其质量,再逆转录获取cDNA。建议实验室设置阳性对照与阴性对照。
Ø 引物和探针的设计:参见4.荧光定量引物设计原则
Ø 选择优良试剂:根据实验目的选择染料法荧光定量检测或探针法荧光定量检测;为了减少操作错误,建议选择2×预混试剂;为了降低非特异性扩增,最好选择Hot Start DNA聚合酶的预混试剂,但需在PCR条件的最初步骤设置10~15分钟的热变性程序,以恢复DNA聚合酶的活性;选用试剂耗材需与荧光定量检测仪配套;选择质量较高的标准品制备标准曲线。
Ø 反应条件的优化:可根据使用的产品说明书进行反应条件的优化。
Ø 实验操作:必须分区操作,避免污染。
9. 扩增曲线的荧光信号值低的原因
扩增曲线存在问题,如扩增曲线的荧光信号值低,通常是由于背景荧光信号值过高造成的。染料法进行检测时,背景荧光信号偏高大多数是由于模板量过高;荧光探针法进行检测时,背景荧光信号偏高大多是因设计的探针质量差
Ø 确认基线或ROX校正前的原始曲线。
Ø 调整模板使用量。
10. 扩增曲线出现朝右下方下落的原因
Ø 使用的模板量过多,扩增曲线过早起峰,使基线校正不能正常进行。建议优化模板使用量。
Ø 基线校正不恰当,确认设置的基线校正范围的参数。
11. PCR扩增效率低的原因
Ø PCR阻害物质的混入,需重新优化获取模板的方法。
Ø 标准品稀释不准确,需使用专用标准品稀释液。
Ø PCR反应条件不佳。引物、试剂、PCR反应条件等优化。
12. 融解曲线出现复数峰的原因
可能是目的片段以外的扩增或引物二聚体的产生
Ø 存在目的基因的Variants,需重新设计引物。
Ø 引物二聚体等非特异性扩增,优化PCR条件或重新设计引物。
Ø 基因组DNA的扩增,需用DNase I处理或考虑重新设计引物。
注:建议对初次使用引物的扩增产物进行电泳确认:单一峰型时可以确认扩增片段大小,判断是否为目的产物;非单一峰型时也有必要对扩增产物进行确认,如果电泳结果显示为单一条带,并与目的片段大小吻合,可以判断为进行了特异性扩增。
13. Ct值的计算方法
Ø 交点法,通过荧光阈值和扩增曲线的交点获得Ct值。
Ø 二次求导法,通过扩增曲线二次求导后取其最大值获取Ct值,其不会随荧光阈值的设定不同而变化,重现性高。
14. RT-qPCR预混试剂有絮状沉淀物的处理方法
白色絮状沉淀物的产生有时是由组分溶解不彻底导致。此时请用手稍微加热或室温短时间放置制品,然后进行上下颠倒或低速短暂涡旋混合,使组分充分溶解,但请不要用振荡器等剧烈搅拌混合。溶解后的试剂性能不受影响。
15. 使用qPCR试剂时,PCR程序选择两步法还是三步法
普通PCR反应程序按三步法进行,而qPCR系列产品的大量实验数据表明,两步法PCR可以减少反应时间,增强反应特异性,提高扩增效率,得到较好的实验结果,推荐使用标准程序两步法PCR。即退火/延伸温度为60℃,可在60~66℃范围进行优化。但需要注意避免退火/延伸设定温度高于68℃,温度过高引物退火不完全,降低扩增效率。如果两步法PCR反应性能较差时,可使用三步法PCR程序进行优化。
免责声明:本站部份内容由优秀作者和原创用户编辑投稿,本站仅提供存储服务,不拥有所有权,不承担法律责任。若涉嫌侵权/违法的,请反馈,一经查实立刻删除内容。本文内容由快快网络小若创作整理编辑!